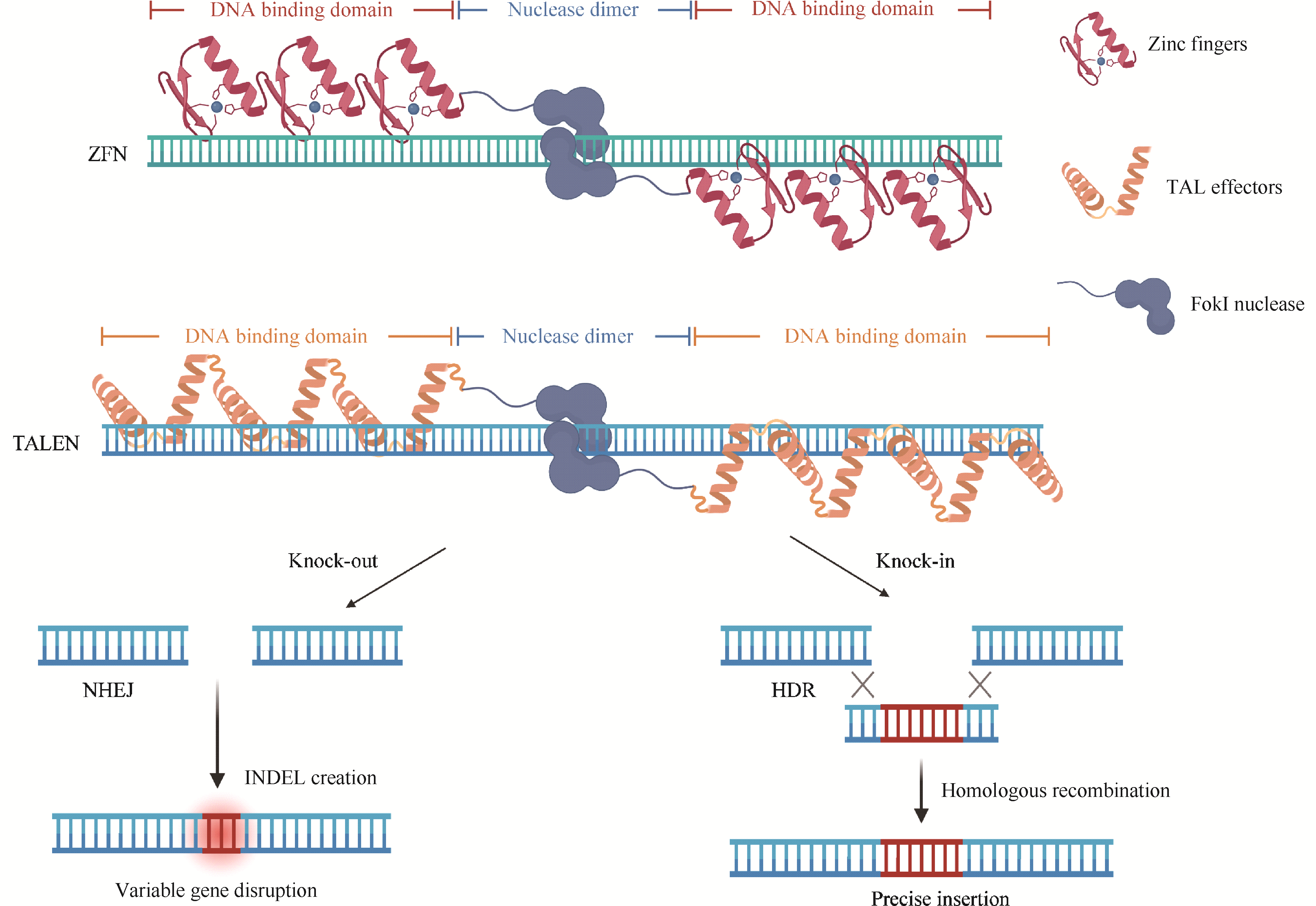
Due to its transparent embryos, rapid external development, high fecundity, and genome highly similar to humans, zebrafish has become an important vertebrate model in biological research. To study gene function, various genetic research methods in zebrafish have been developed, primarily including forward and reverse genetic approaches. Forward genetics starts from observable phenotypes and identifies the causal genes by screening mutants with specific traits, whereas reverse genetics starts from known genes and investigates their function by altering their sequence or expression level. Together, these two strategies constitute an important theoretical foundation for functional genomics research in zebrafish. Reverse genetics focuses on one or more specific genes to investigate the relationship from genotype to phenotype, and it plays an increasingly important role in zebrafish genetic research. Reverse genetic techniques in zebrafish mainly include gene overexpression, gene downregulation, and gene knockout, which enable the manipulation of specific genes to study their critical roles in physiological development and pathological processes. The development of these techniques has significantly advanced genetic research in zebrafish and facilitated the establishment of various zebrafish models for human diseases, contributing substantially to the understanding of disease mechanisms. This article will provide a detailed review of the research progress in reverse genetic techniques in zebrafish and discuss future perspectives.
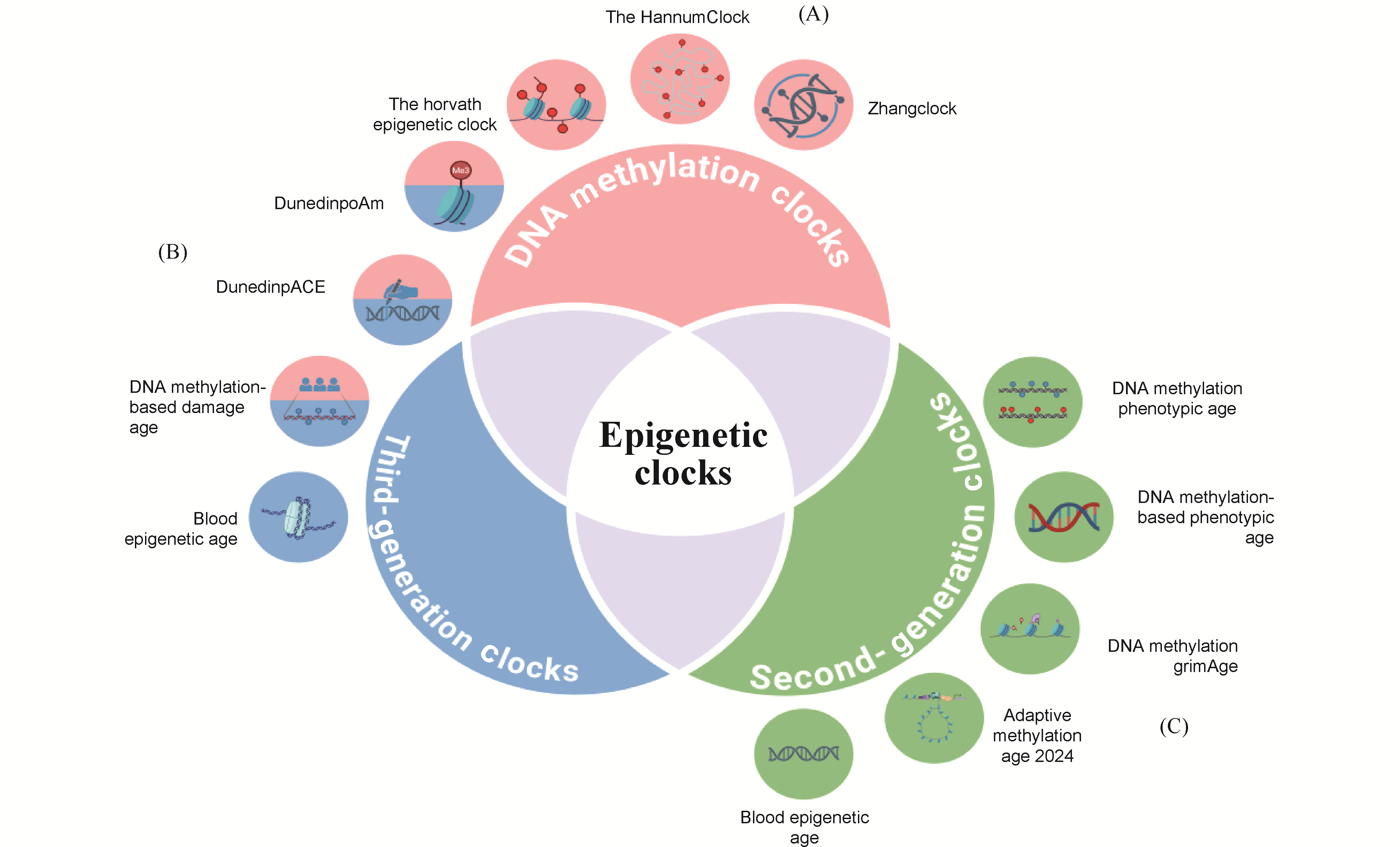
Epigenetic clocks estimate biological age by analyzing patterns of epigenetic marks; among them, DNA-methylation-based clocks are the most mature. This review systematically outlines their recent advances in precision medicine. First-generation clocks rely on linear-regression models and a modest number of CpG sites, offering cross-tissue applicability yet limited accuracy. Second-generation clocks incorporate multi-tissue features and advanced statistics, markedly improving disease-risk prediction. Third-generation clocks leverage deep learning on genome-wide methylation maps, delivering higher precision and broader population generalizability. Current challenges include pronounced data heterogeneity, limited model portability, high clinical-translation costs, and ethical concerns. Countermeasures should center on standardizing detection protocols, assembling multi-ethnic datasets, developing low-cost technologies, and refining ethical oversight. By quantifying biological-age acceleration, DNA-methylation clocks provide novel tools for chronic-disease risk prediction, personalized interventions, and health management, poised to accelerate the clinical adoption of precision medicine in aging and related disorders.
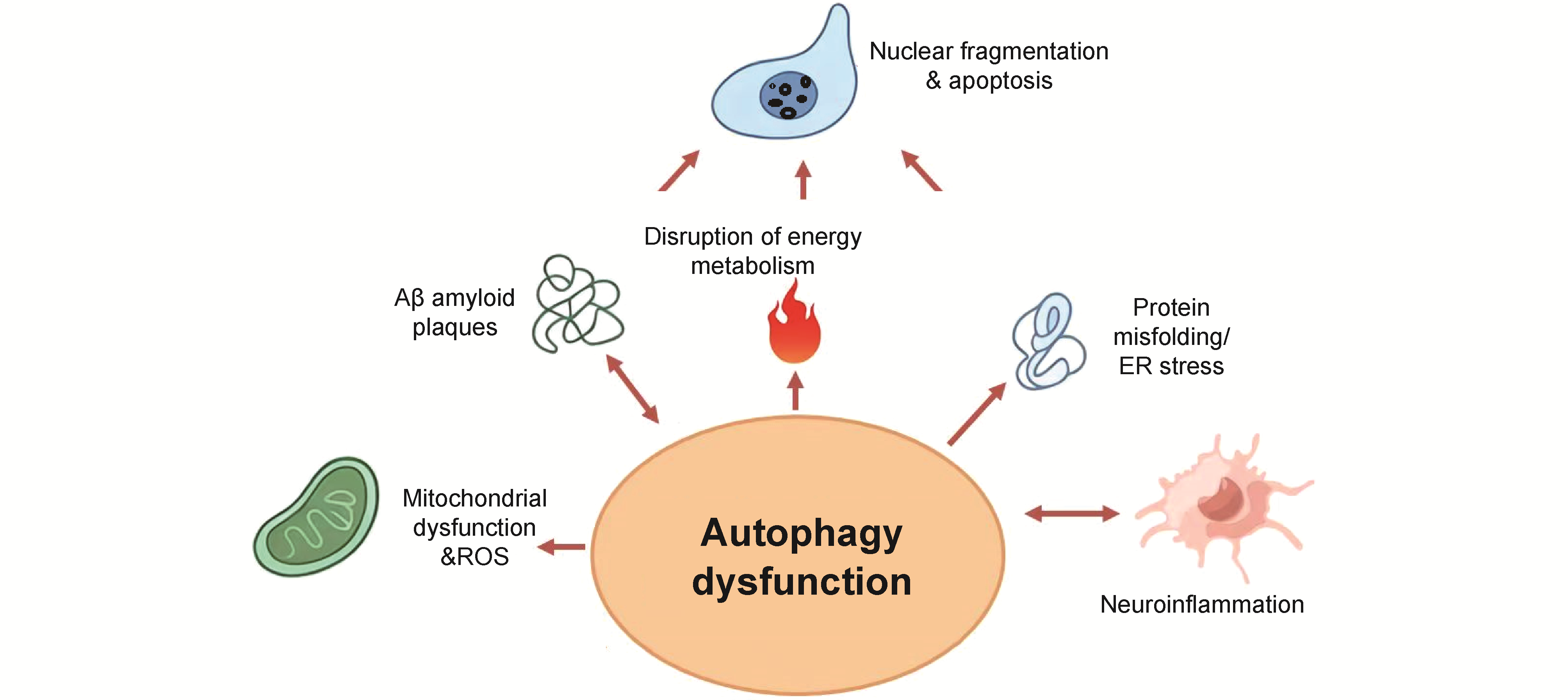
Neurodegenerative diseases (NDDs) such as Alzheimer's disease are a class of chronic progressive diseases characterized by neuronal damage and death. With the aging of the global population, the incidence of such diseases is increasing year by year, and they are currently lacking effective cures. Autophagy can be deeply involved in the disease processes of NDDs by regulating key links, such as the accumulation of neurotoxic proteins, neuroinflammation, oxidative stress, energy metabolism and lactylation. This paper begins by discussing the mechanisms and processes of autophagy, then summarizes the pathological mechanisms of NDDs and their relationship with autophagy, with a focus on recent research advances in the regulatory role of autophagy in the pathological progression of NDDs. Finally, we explore the potential value of autophagy intervention in the treatment of NDDs. This paper also provides an in-depth analysis of existing challenges in current research. According to available evidences, pathological processes related to NDDs generally activate autophagy yet often lead to impairments in its normal function. Autophagy can degrade pathological aggregates such as amyloid-β and α-synuclein through lysosome-related mechanisms, alleviate neuroinflammation by clearing damaged mitochondria and suppressing microglial overactivation, and reduce cellular damage caused by energy metabolism disorders through the removal of dysfunctional mitochondria. Conversely, autophagy dysfunction results in increased accumulation of toxic aggregates, elevated release of pro-inflammatory cytokines, impaired mitochondrial function, induced oxidative stress, and disrupted cellular energy metabolism. The increased lactylation modification caused by the above pathological processes further leads to the disorder of autophagic function and the exacerbation of pathological processes such as neuroinflammation. Drugs with autophagy-regulating functions, such as oleanolic acid, have already demonstrated certain potential in the treatment of NDDs.
Helium, the second most abundant element in the universe and an important member of the noble gas group, has been widely used in aerospace, medical imaging, and superconductivity, due to its unique properties of low density, high thermal conductivity, and chemical inertness. With the in-depth study, scientists found that this chemically stable gas could regulate the physiological and pathological processes. In medical research, for example, animal studies demonstrated that helium has significant organ-protective and neuroprotective effects. In agricultural studies, helium effectively extends the shelf life of agricultural products and enhances crop tolerance upon environmental stimuli. These findings clearly suggested that helium might exert its protective effects by modulating specific pathways influencing the physiological metabolism and responses. To better understand the research achievement of helium-based biology, the discovery of helium, and helium-based biological effects in animal models, agricultural products preservation, and crop tolerance against stress from both medical and agricultural perspectives were reviewed, and we also summarized its potential molecular mechanisms, which were functionally associated with the regulation of mitochondrial function, activation of anti-apoptotic pathways, inhibition of oxidative stress, induction of nitric oxide signaling pathways, reduction of respiration rate and ethylene release, suppression of microbial growth, as well as the reestablishment of redox balance and ion homeostasis. Finally, the current limitations and potential future research directions were outlined, aiming to offer a solid reference for helium-based biology research and possible applications.
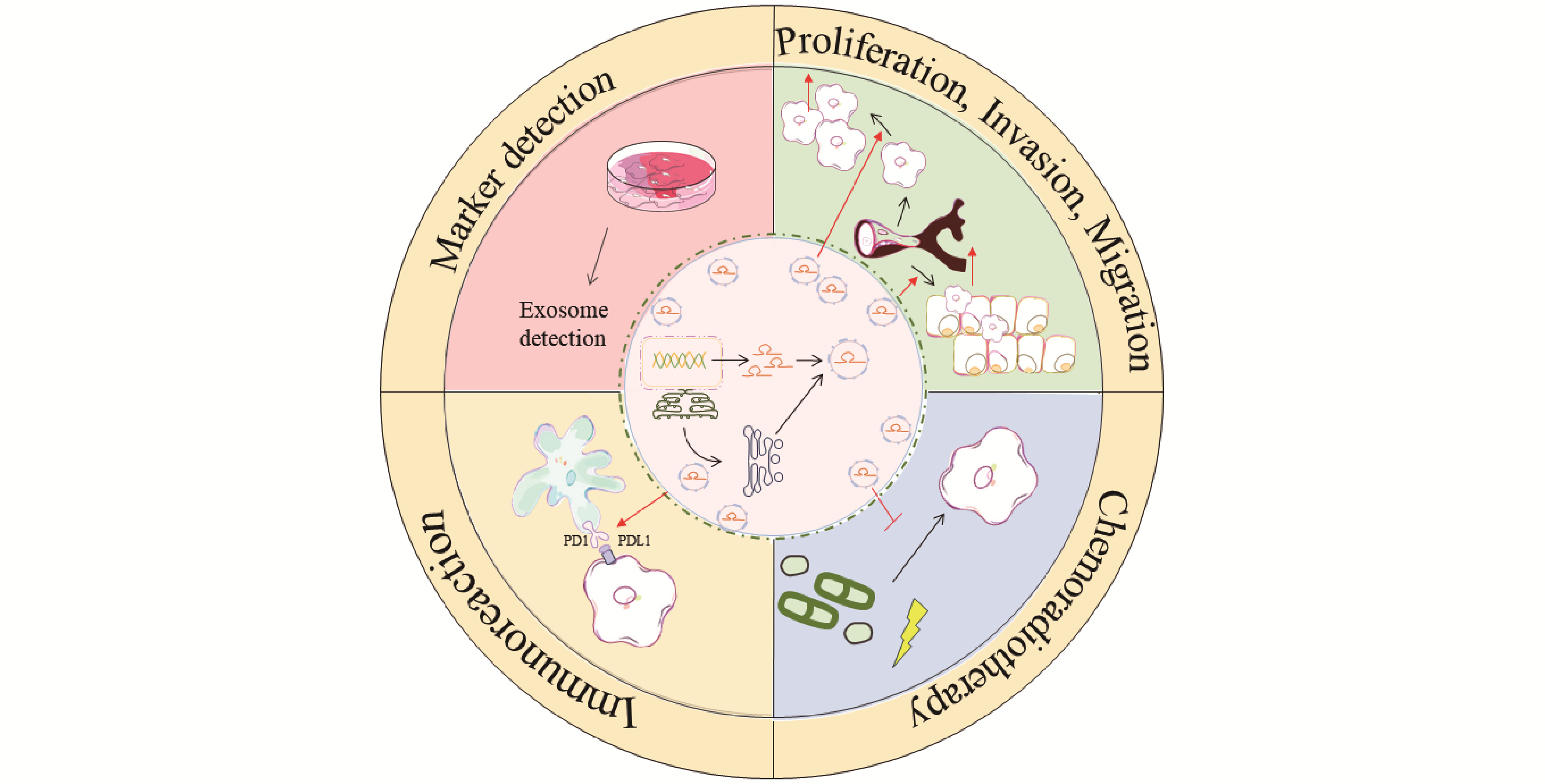
Esophageal squamous cell carcinoma (ESCC) is a highly malignant gastrointestinal cancer with a poor prognosis. Due to its insidious early symptoms, difficulties in diagnosis, and limited efficacy of current treatment strategies, the five-year survival rate of ESCC patients remains low. In recent years, exosomes have attracted considerable research interest as key mediators of intercellular communication. They carry a variety of bioactive molecules, including proteins, lipids, and nucleic acids. Among these, long non-coding RNAs (lncRNAs) play a crucial role as functional RNAs and are central to the pathogenesis, invasion, metastasis, and drug resistance of ESCC. Studies have shown that exosomal lncRNAs participate in the initiation and progression of ESCC through multiple mechanisms. On the one hand, they can be secreted by tumor cells and cancer-associated fibroblasts, and then delivered to recipient tumor cells, where they regulate key signaling pathways that influence tumor cell proliferation, invasion, and migration. On the other hand, exosomal lncRNAs also mediate immune regulation and confer radio- and chemoresistance within the tumor microenvironment of ESCC. Particularly noteworthy is the fact that exosomal lncRNAs are widely present in body fluids such as blood and saliva, characterized by high stability and ease of enrichment and detection, making them ideal biomarkers for non-invasive early screening, dynamic therapeutic monitoring, and personalized prognostic evaluation in ESCC. This article systematically reviews the molecular mechanisms by which exosomal lncRNAs regulate ESCC progression, providing an in-depth discussion of their clinical translational potential and application prospects as novel diagnostic markers and precision therapeutic targets.
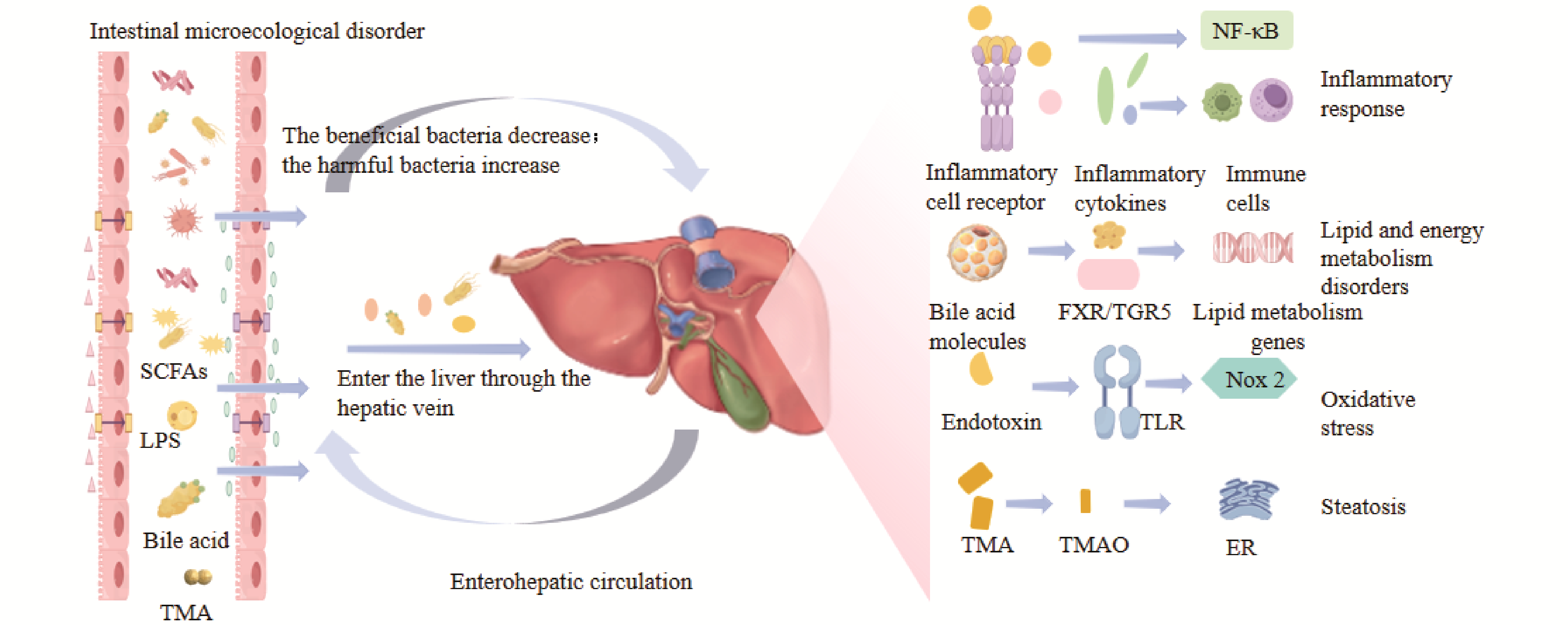
Non-alcoholic fatty liver disease (NAFLD) is one of the most prevalent chronic liver diseases worldwide, characterized as a clinicopathological syndrome primarily by diffuse macrovesicular steatosis of hepatocytes. In recent years, studies have indicated that intestinal flora dysbiosis is closely associated with the occurrence and progression of NAFLD. With the increase in the number of harmful bacteria, they inhibit beneficial bacteria. Beyond disrupting normal lipid metabolism, microecological imbalance can also impair the integrity of the intestinal mucosal barrier, leading to increased intestinal permeability. Endotoxins produced by the metabolism of harmful bacteria enter the liver and trigger persistent inflammatory responses, which is particularly detrimental to liver physiology. In contrast, gut microbiota reconstruction can intervene in the disease process by promoting hepatic lipid metabolism, restoring the integrity of the intestinal barrier, and inhibiting inflammation. Traditional Chinese medicine (TCM) exerts its advantages of multi-pathway, multi-target, and mild adverse reactions by increasing the abundance of beneficial bacteria and reducing that of harmful bacteria. This thereby reduces fat accumulation, promotes the restoration of intestinal barrier integrity, and inhibits hepatic inflammation, further achieving holistic regulation of patients and providing a new model for the diagnosis and treatment of NAFLD.
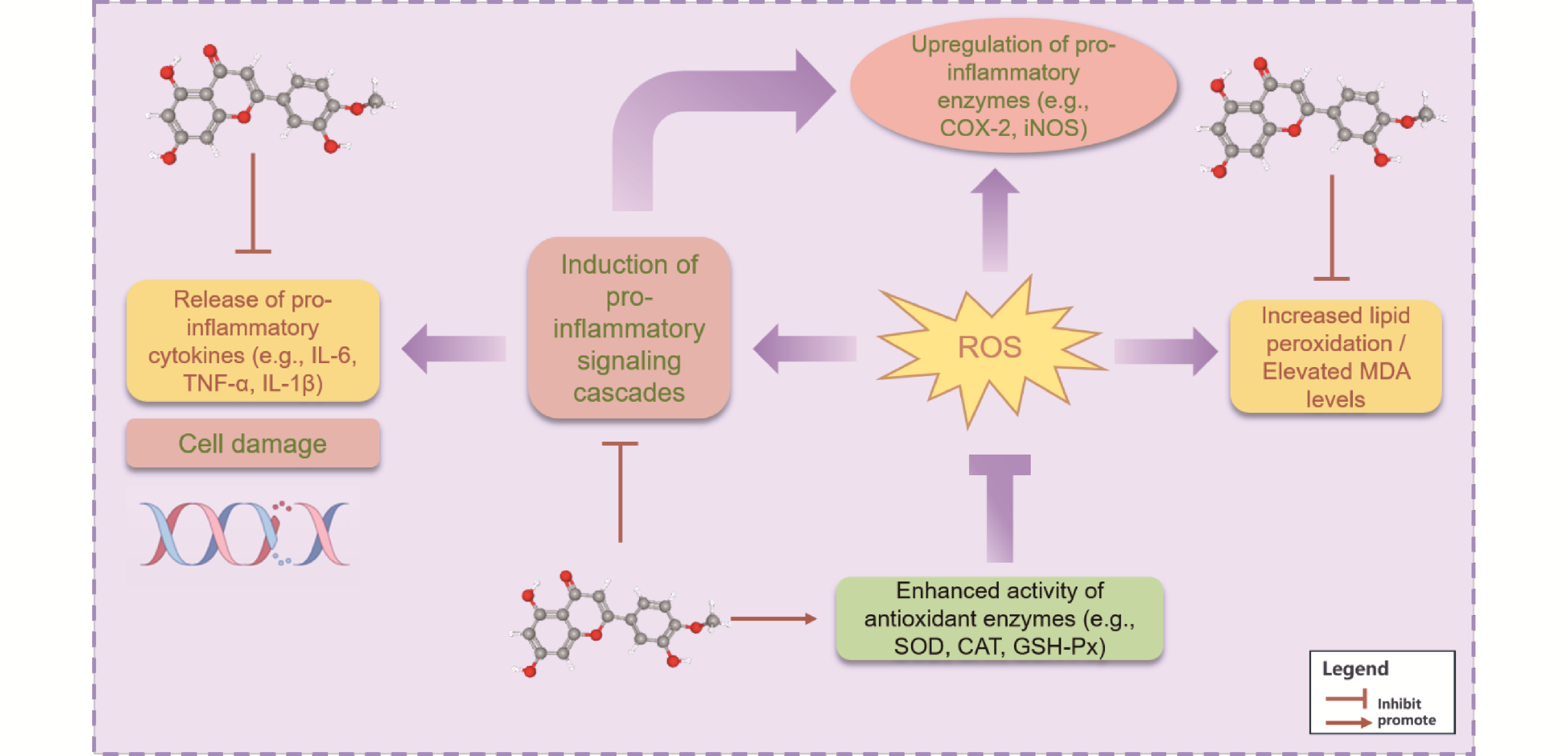
Chronic inflammation is a key pathogenic factor in numerous debilitating diseases, including arthritis, neurodegenerative disorders, and metabolic syndrome. Conventional therapies such as NSAIDs often provide symptomatic relief but are limited by adverse effects and the development of resistance. Consequently, significant research has focused on natural therapeutic agents. Diosmetin, a natural flavonoid abundant in citrus peels, olive leaves, and vetch, exhibits potent anti-inflammatory and antioxidant activities. This review elucidates the molecular mechanisms of diosmetin, detailing its systemic regulation of complex signaling networks that govern the inflammatory response. Its core mechanism involves the dual suppression of pro-inflammatory pathways, including NF-κB, MAPK, and JAK/STAT, thereby reducing the expression of inflammatory mediators. Concurrently, it activates the protective Nrf2 antioxidant pathway, establishing an efficient regulatory network that suppresses inflammation while promoting cellular defense. Furthermore, this review discusses emerging strategies, such as advanced nano-formulations, to overcome its characteristically low bioavailability and enhance its clinical potential. This work provides a comprehensive theoretical framework to guide the development of novel diosmetin-based therapeutics for managing chronic inflammatory diseases.
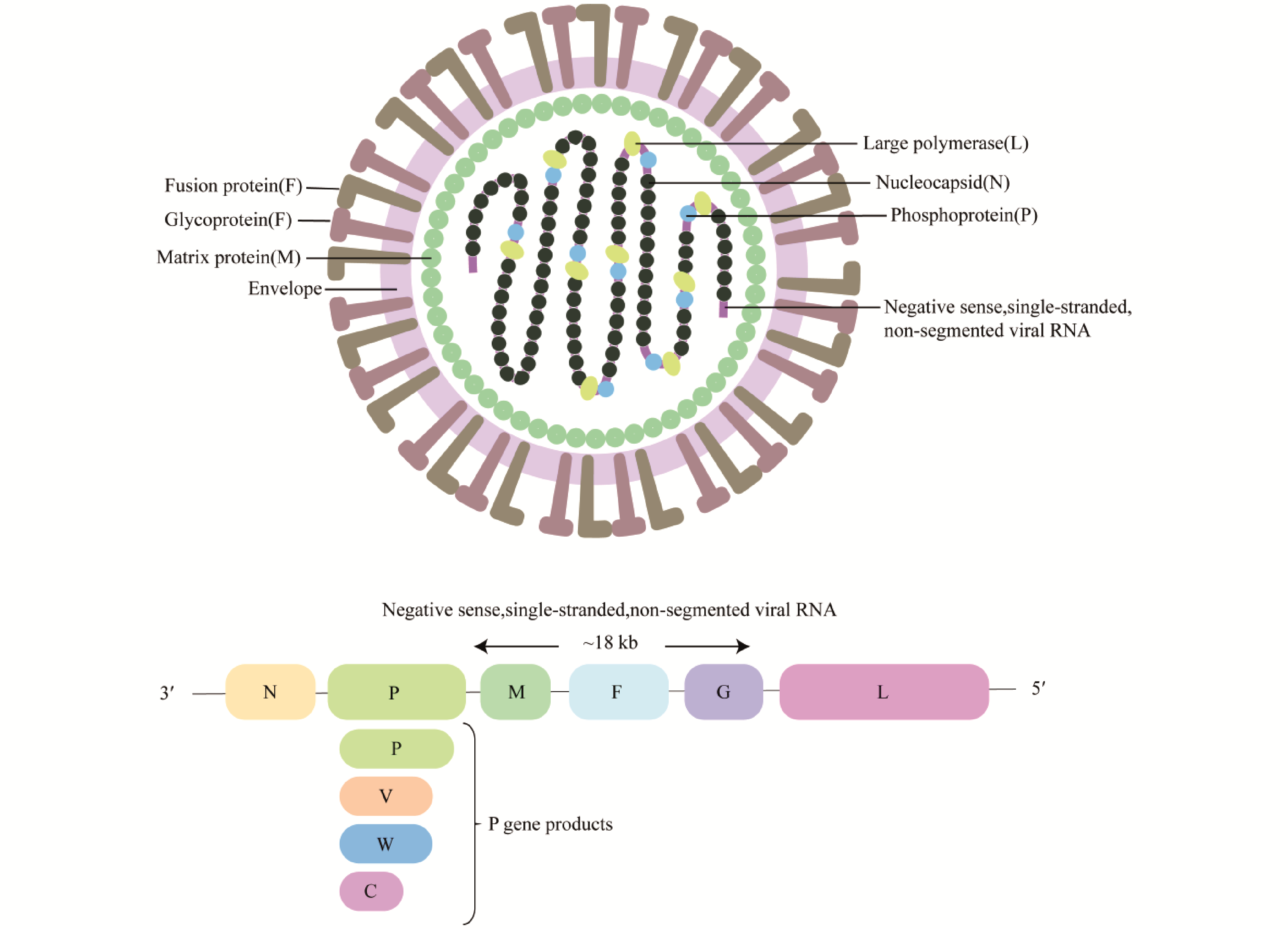
Nipah virus (NiV) is a zoonotic virus, which has become a major threat to global public health due to its cross-species transmission ability and high mortality rate. Fusion protein (F) is the core molecule of NiV entering host cells and plays a key role in viral infection and immune escape. It is not only the structural basis of viral invasion, but also a key immune target. In recent years, the research on the structural stability, epitope distribution and fusion activation mechanism of F protein has been in-depth, which has promoted the rapid development of various vaccine platforms. This paper focuses on the structural characteristics of F protein, especially the structural division and functional synergy of F1 and F2 subunits in virus-host membrane fusion, and emphasizes its important role in inducing neutralizing antibodies and stimulating protective immune responses. In addition, a variety of vaccine platforms based on F protein, including viral vectors, DNA, mRNA and virus-like particle (VLPs) vaccines, are reviewed, and the correlation of immune protection is discussed by combining experimental data from different animal models. Based on the current research progress, the core value of F protein in analyzing the mechanism of NiV infection and promoting vaccine development was highlighted, providing a theoretical basis for clinical translation and public health prevention and control.
Aspartylglucosaminidase (AGA) is an amidohydrolase that catalyzes the hydrolysis of the amide bond between N-acetylglucosamine (GlcNAc) and asparagine (Asn), yielding N-acetylglucosamine and aspartic acid (Asp) as products. Belonging to the N-terminal nucleophile hydrolase (Ntn hydrolase) family, AGA is synthesized as a nascent polypeptide chain that undergoes sequential autohydrolysis to form α and β subunits, which then assemble into an α2β2 heterotetramer. Its "funnel-shaped" active domain provides a reaction site for substrate binding and catalysis. AGA is widely distributed across various organisms, including humans, mice, cattle, fungi, and bacteria. As one of the key enzymes involved in the intracellular degradation of N-glycoproteins, the cleavage products of AGA can participate in substance synthesis or energy metabolism, thereby maintaining intracellular metabolic homeostasis. Additionally, AGA plays a crucial role in regulating the development of the central nervous system and motor function in mice. Research has indicated that parasitic wasps can utilize AGA present in their venom to manipulate the development of Drosophila hosts, ensuring the survival of their own offspring. Mutations in the human AGA gene lead to aspartylglycosaminuria (AGU), a rare hereditary disorder. The main clinical manifestations of AGU include developmental delay, intellectual disability, abnormal skeletal development, and abnormal connective tissue growth. Diagnosis of this disease relies on AGA enzyme activity assay, genetic testing, and urine GlcNAc-Asn screening. Currently, therapeutic approaches for AGU remain in the research stage. While certain progress has been made in research on gene therapy (e.g., delivery of human AGA via AAV9 vector), enzyme replacement therapy, and chaperone therapy (e.g., betaine-facilitated folding of AGA mutants), further research and validation are still required prior to their clinical translation and application. This review summarizes the distribution of AGA across different species, its three-dimensional structure, enzymological functions, biological functions, and its association with diseases, aiming to provide a basis for the basic and applied research on AGA.
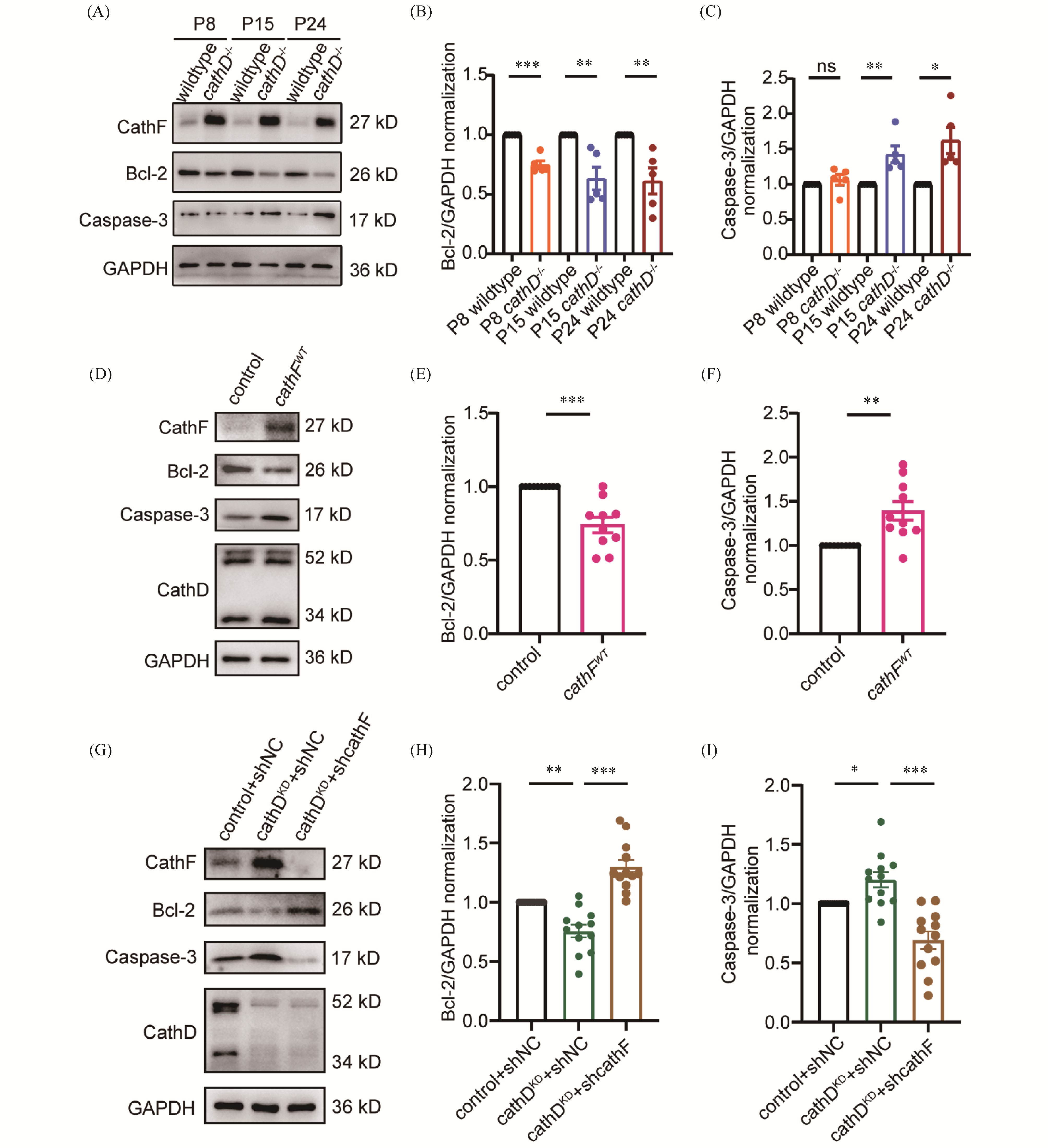
Cathepsin D (cathD), an aspartic protease in lysosomes, is involved in regulating cell apoptosis, differentiation and cell cycle. Mutations of cathD gene can result in neuronal ceroid lipofuscinosis (NCL), a lysosomal storage disease (LSD), and patients with this disease often suffer from sensorineural hearing loss (SNHL), but the mechanism of deafness has not been fully elucidated. Therefore, to explore the regulatory role of cathD in sensorineural hearing loss, this study constructed cathD knockout mice (cathD-/-) to detect the morphological development and hearing function of the cochlea at different developmental stages in mice, trying to clarify the regulatory mechanism of cathD in the cochlea of mice. In this study, auditory brainstem response (ABR) was used to test the hearing function of cathD-/- mice, and it was found that the knockout of cathD gene caused hearing loss in mice (P<0.01). Immunofluorescence staining and scanning electron microscopy observation suggested the loss and morphological abnormalities of cochlear hair cells (P<0.01). In addition, proteomics analysis was used to detect the cochlear proteins of cathD-/- mice at different developmental stages, and it was found that the expression of cathepsin F (cathF) was abnormally elevated (P<0.01). Westernblot analysis showed that the expression of Bcl-2 in cochlear tissue of cathD-/- mice was decreased (P<0.01) and the expression of cleaved caspase-3 was increased (P<0.05). Furthermore, this result was further validated through cellular experiments targeting the cochlear hair cell line. These results suggested that after cathD gene knocking-out, it can induce cell apoptosis by increasing the expression of cathF, inhibiting the expression of Bcl-2, and activating the caspase-3 mediated mitochondrial apoptotic pathway, thereby affecting the morphology of cochlear hair cells and hearing function in mice.
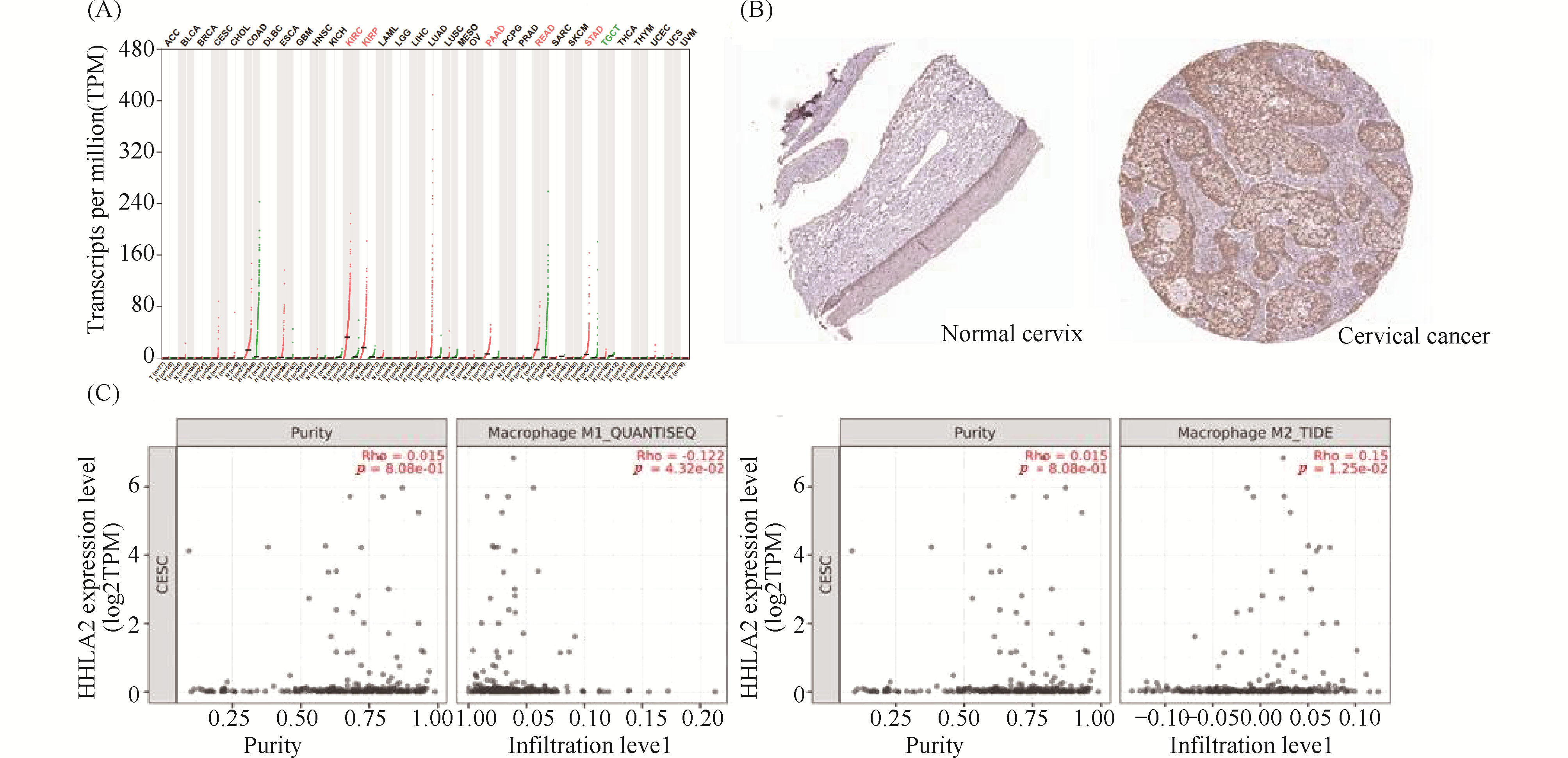
Long non-coding RNAs (lncRNAs) play critical roles in tumorigenesis and regulation of the immune microenvironment, yet their mechanisms in cervical cancer progression remain unclear. In this study, we performed whole-transcriptome RNA sequencing on samples from control, high-grade squamous intraepithelial lesion (HSIL), and cervical squamous cell carcinoma (CESC) groups. By integrating differential expression analysis with machine learning approaches, we identified key lncRNAs and constructed a competing endogenous RNA (ceRNA) regulatory network, highlighting the lncRNA NONHSAT001931. Further analysis revealed a potential regulatory axis involving NONHSAT001931, miR-4699-5p, and HHLA2. Database validation and immunohistochemistry demonstrated that HHLA2 is highly expressed in cervical cancer tissues and its expression correlates with M1/M2 macrophage infiltration, suggesting that this axis may contribute to the formation of an immunosuppressive tumor microenvironment. Overall, our findings uncover the potential role of this ceRNA axis in the progression from HSIL to cervical cancer, providing a theoretical basis for the investigation of immune mechanisms and therapeutic targets in cervical cancer.
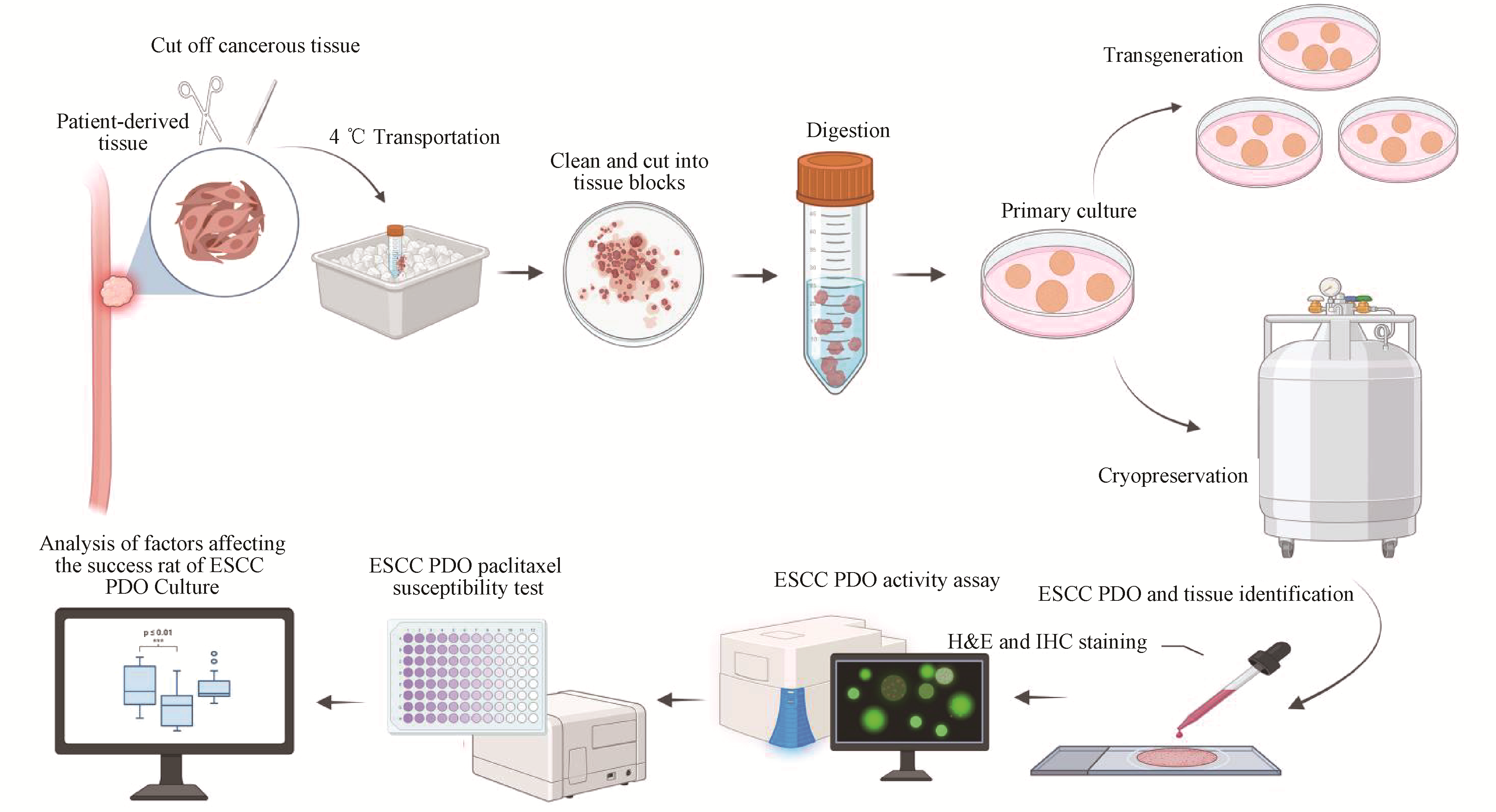
Esophageal squamous cell carcinoma (ESCC) is characterized by high invasiveness and poor prognosis, with distinct geographical distribution patterns. Research progress has long been hampered by the lack of suitable preclinical models. Patient-derived organoids (PDO) can more accurately recapitulate the tissue architecture and functional characteristics of human organs compared to 2D cell cultures and animal models. In this study, we established a stable patient-derived esophageal squamous cell carcinoma organoid (ESCC PDO) culture platform using clinical samples from a high-incidence area, achieving a success rate of 68.0%. Hematoxylin-eosin (H&E) staining and immunohistochemistry (IHC) confirmed that the PDO models closely retained the pathological features of the original ESCC tissues. Calcein/PI double staining demonstrated that ESCC PDOs maintained good cell viability at various stages. Drug sensitivity assays using PDOs effectively recapitulated the inter-individual variability in patient drug responses. Chi-square test and univariate logistic regression analysis revealed that tumor differentiation (P=0.03), T stage (P=0.01), neoadjuvant chemotherapy outcome (P=0.03), time of tumor tissue ex vivo (P=0.00118), and tumor weight (P=0.000273) were significantly correlated with the success rate of ESCC PDO culture. In contrast, patient age, gender, N stage, and tumor location showed no significant correlation with the culture success rate (P>0.05). This study identifies key factors influencing the successful establishment of ESCC PDOs, and provides a more precise in vitro model for ESCC research in high-incidence regions.
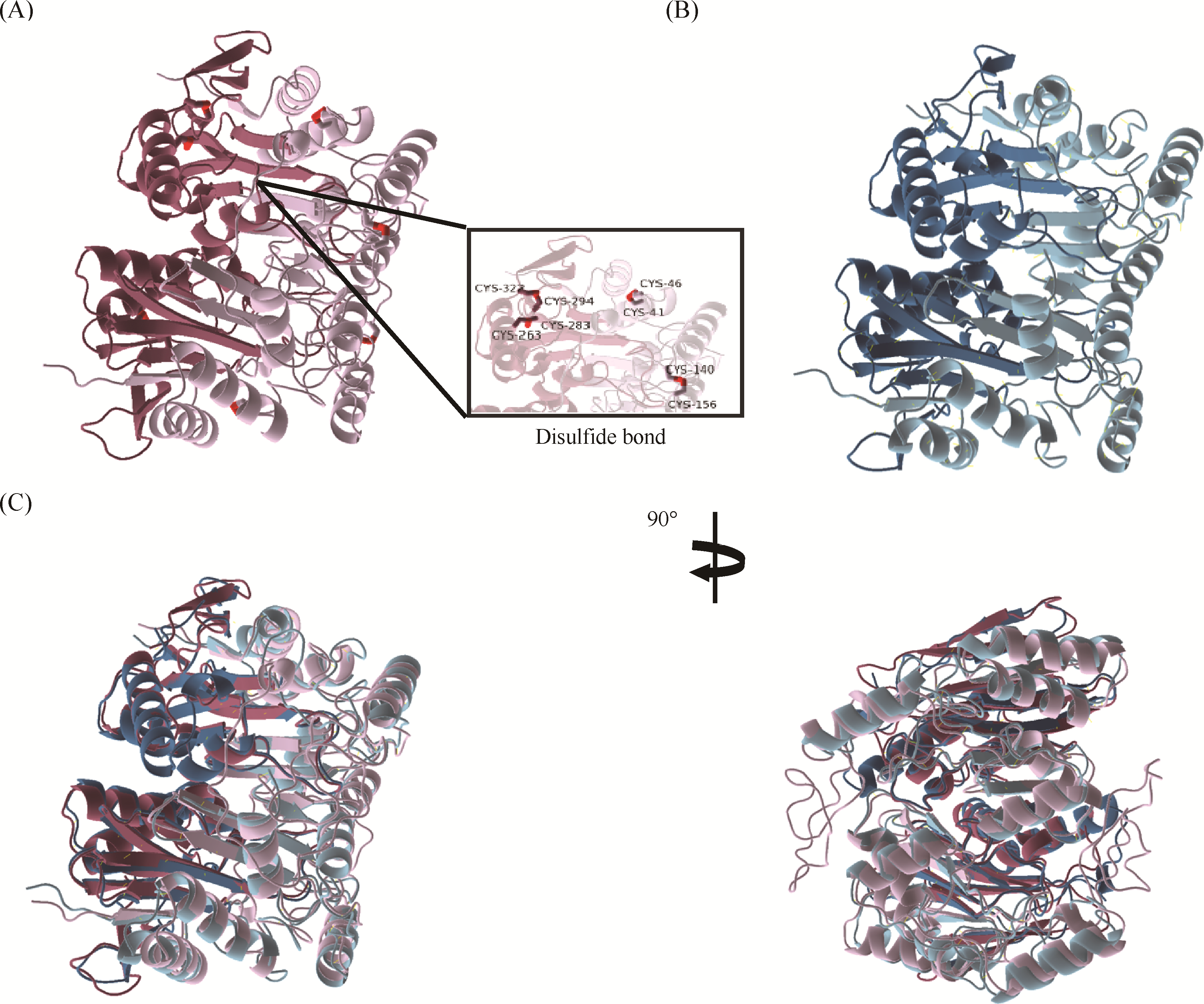
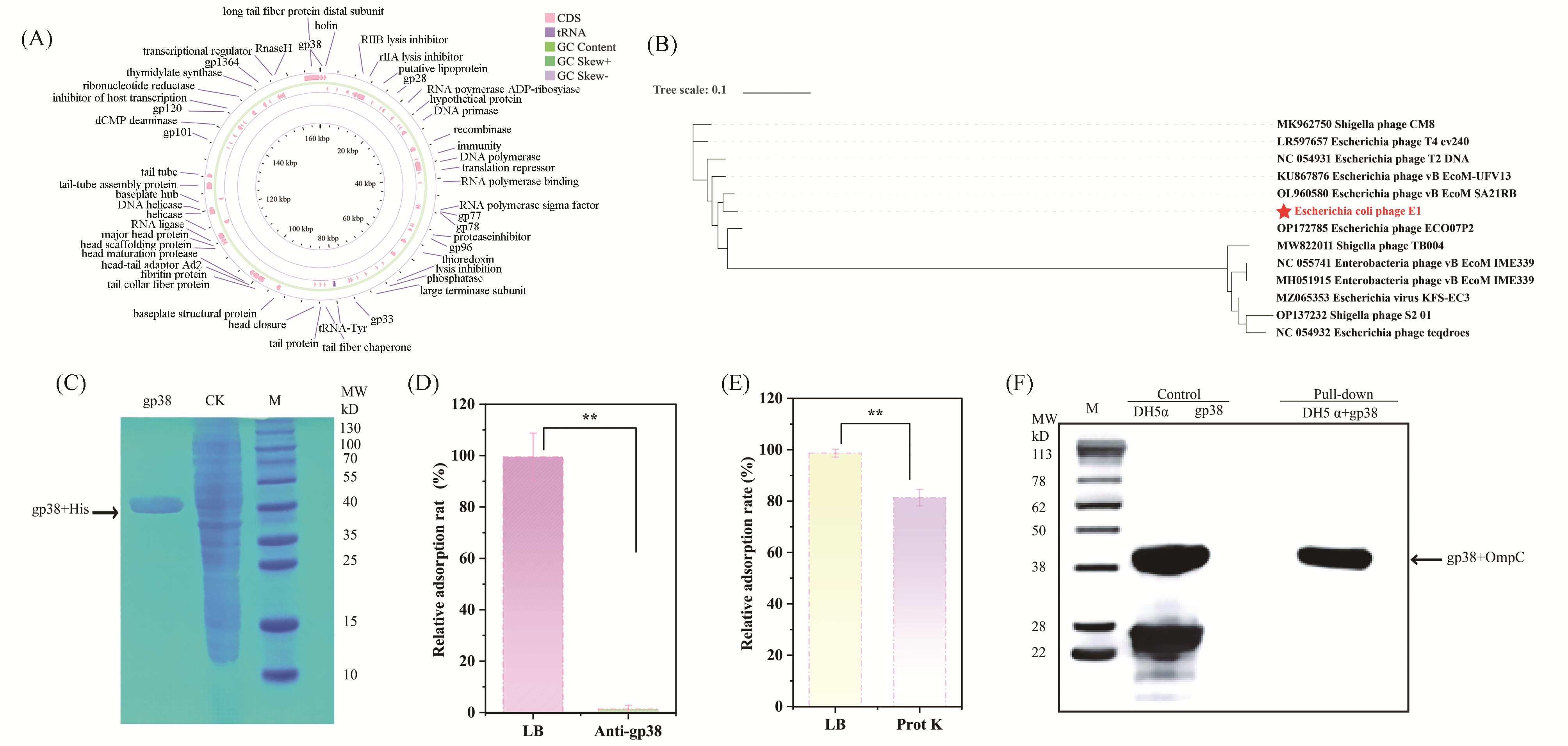
Multidrug-resistant bacterial infections represent a major cause of mortality worldwide, underscoring the urgent need for alternative antibacterial strategies, among which bacteriophage therapy has emerged as a promising approach. A detailed understanding of phage-host recognition is fundamental to the development of precision phage therapeutics. In this study, we investigated a newly isolated virulent T2-like bacteriophage, E1, and dissected the molecular mechanism underlying the interaction between its receptor-binding protein (RBP) and the host receptor. Through whole-genome sequencing and phylogenetic analysis, the terminal tail fiber protein gp38 was identified as a putative RBP. Antibody-blocking assays subsequently confirmed gp38 as the functional RBP of phage E1. Furthermore, host surface protease digestion, combined with in vitro pull-down assays and Western blotting analysis, established the Escherichia coli outer membrane protein C (OmpC) as the specific host receptor. To resolve the recognition mechanism at the molecular level, an E1 gp38-OmpC complex model was generated using AlphaFold3, followed by 500-ns molecular dynamics simulations. The results indicate that complex formation is primarily driven by van der Waals and electrostatic interactions. Alanine scanning energy analysis identified a cluster of critical energetic hotspots composed of Trp9, Arg21, Trp22, and Trp37, with Trp22 and Trp37 contributing most prominently to binding (ΔΔG > 12 kcal/mol). Collectively, this study reveals a “lock-and-key” recognition mechanism whereby phage E1 engages the OmpC lumen through a tryptophan/arginine cluster in gp38, and offers molecular insights that may guide the rational engineering of RBPs for precise modulation of the bacteriophage host range.
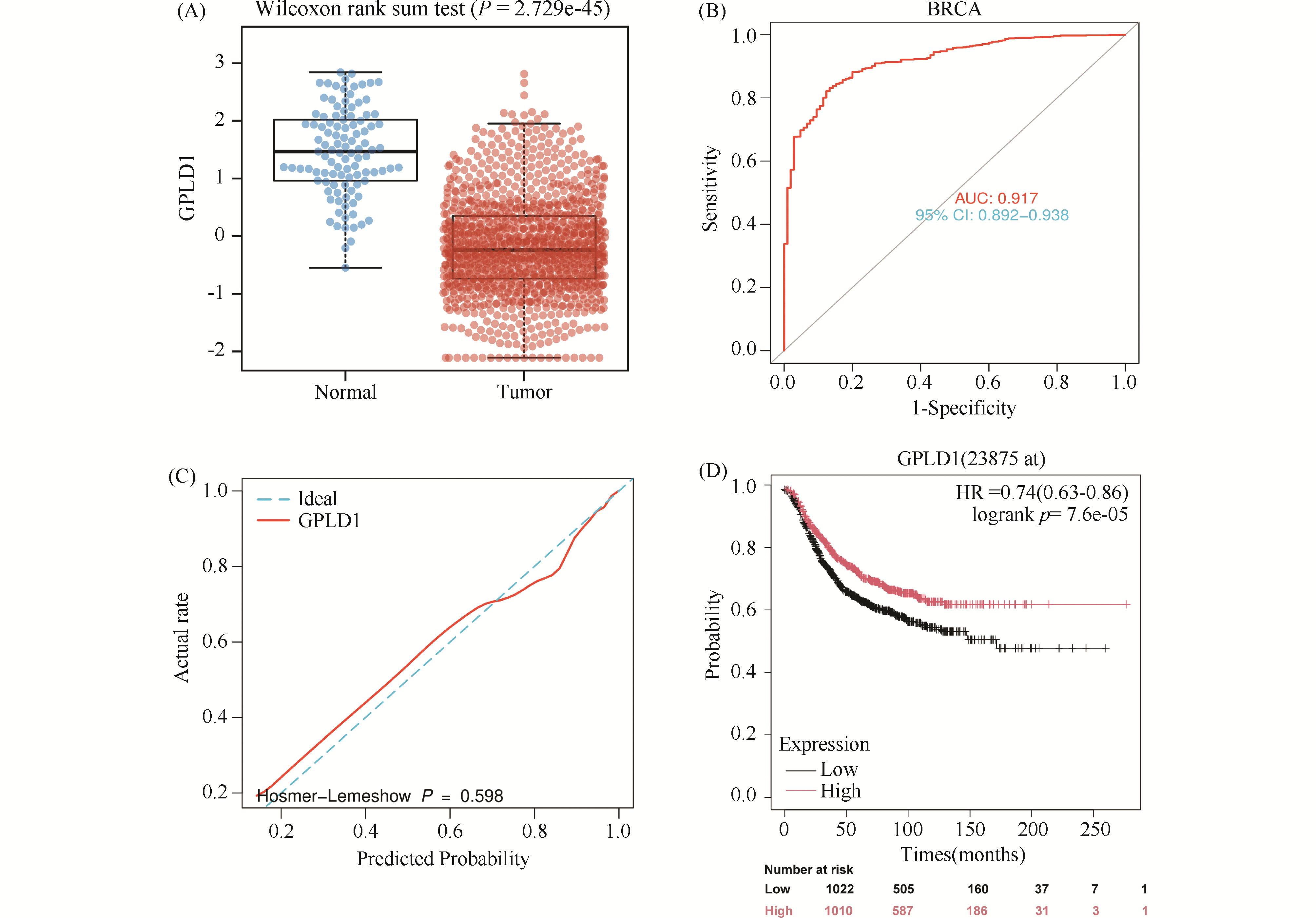
This study aims to investigate the regulatory role and underlying mechanism of glycosy-lphosphatidylinositol-specific phospholipase D1 (GPLD1) in breast cancer progression and to evaluate its clinical diagnostic value. Bioinformatic analysis revealed differential expression of GPLD1 across various tumor types, with significant downregulation at both mRNA and protein levels in breast cancer tissues. This low expression was associated with poor patient prognosis, suggesting GPLD1's potential as a diagnostic biomarker.Survival analysis showed that patients in the GPLD1 high-expression group hadsignificanty better recurrence-free survival (RFS) than those in the low-expression group. suggesting that GPLD1may serve as a potential diagnostic biomarker for breast cancer CCK-8 assay, colony foration assay, Transwellassay and Matrigel Transwell assay in MDA-MB-231 and HS578T cells showed that GPLD1 overexpressioninhibited the breast cancer cells proliferation (P<0.01), migration (P<0.05) and invasion (P<0.01) Flowcytometry and TUNEL staining indicated that up-regulation of GPLD1 facilitated breast cancer cell apoptosis(P<0.05), with no significant effect on cell cycle distribution was observed, Bioinforatics analysis andco-immunoprecipitation (Co-IP) assays confirmed that GPLD1 directly interacts with phosphatidic acidphosphatase 1 (PLPPl). The expression levels of the two proteins were positively correlated in breast cancertissues. and they were co-localized within cells: Nile Red assay demonstrated that concurrent expression ofGPLD1 and PLPP1 exerted a synergistic effect on decreasing intracellular lipid levels (P<0.05); siRNAinterference of PLPP1 could partially restore GPLD1-mediated suppression of cell growh and decreased lipidlevels in breast cancer cells (P<0.01) Collectively, GPLD1 acts as a breast cancer suppressor. It possibly modulates lipid metabolism via interaction with PLPP1 to restrain tumor growth The results of this paper offeringa new candidate target for breast cancer diagnosis and therapy.
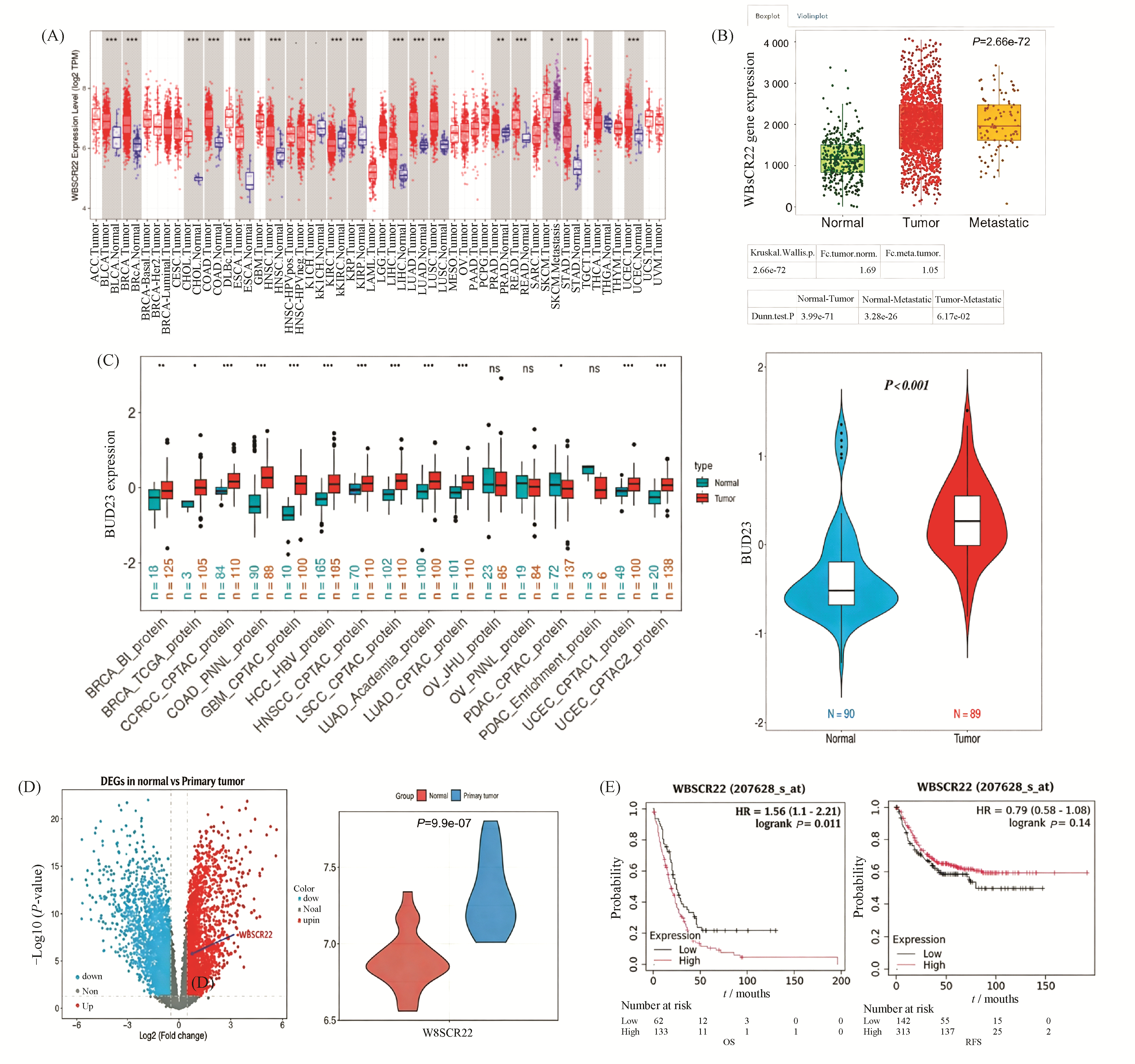
Williams-Beuren syndrome chromosome region 22 (WBSCR22) is an rRNA methyltransferase and plays a role in tumor development. But its mechanism remains poorly understood. Previous studies from our research group found that the expression of WBSCR22 was significantly downregulated in oxaliplatin-resistant colorectal cancer (CRC) cells co-cultured with natural killer (NK) cells. To investigate the function and mechanism of WBSCR22 in CRC, bioinformatics analysis revealed that high expression of WBSCR22 is significantly associated with the overall survival (OS) of CRC patients. Functional experiments demonstrated that overexpression of WBSCR22 promotes the proliferation and migration of CRC cells. Using liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis, SNIP1 was identified as the most significantly altered downstream protein(FC≥1.2,P<0.05). Further studies showed that knockdown of SNIP1 inhibits the proliferation, migration, and in vivo tumor-forming ability of CRC cells, while overexpression of WBSCR22 in SNIP1-knockdown cells partially restores SNIP1 expression and reverses the tumor-suppressive effects caused by SNIP1 deficiency. Mechanistic studies indicated that WBSCR22 may regulate SNIP1 expression by activating the PI3K/Akt/β-catenin signaling pathway. In summary, the potential mechanism by which WBSCR22/SNIP1 promotes CRC cell proliferation, migration, and tumor formation likely involves the PI3K/Akt/β-catenin signaling pathway. We propose that the WBSCR22/SNIP1 axis holds promise as a potential diagnostic biomarker, prognostic indicator, and therapeutic target for CRC.
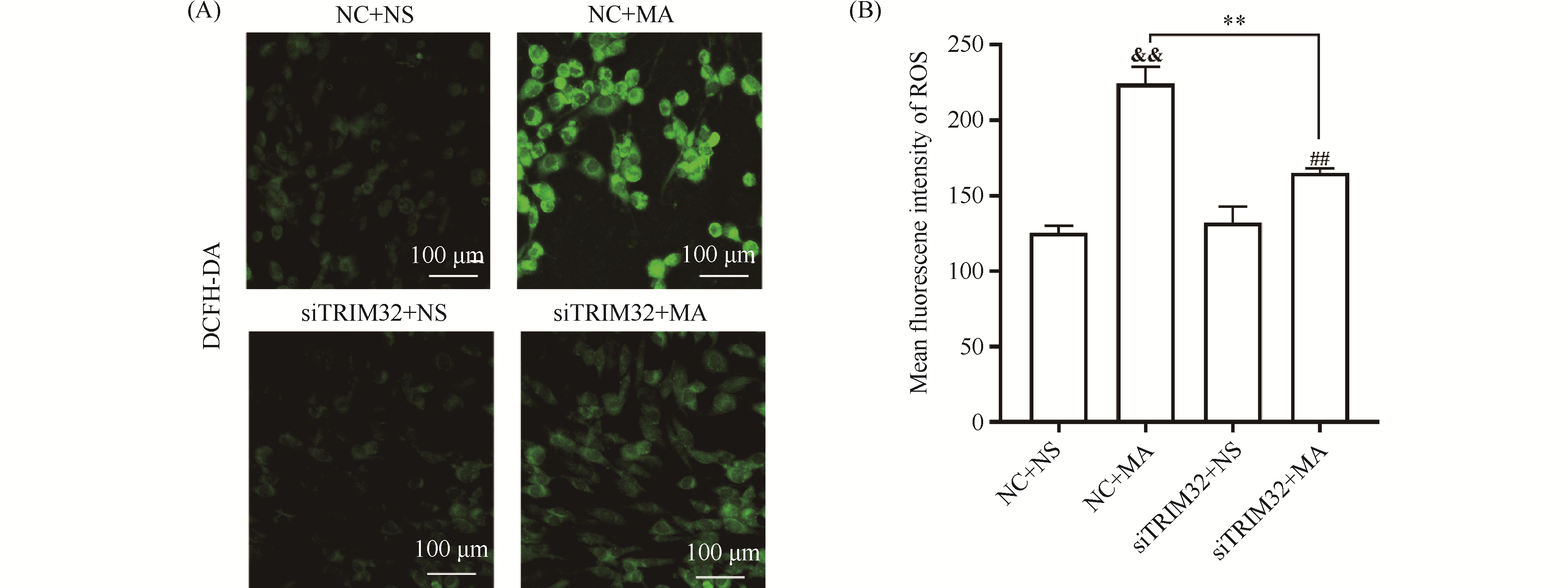
Tripartite motif protein 32 (TRIM32), an E3 ubiquitin ligase, plays pivotal roles in diverse biological processes including myogenesis, tumorigenesis, and metabolic regulation, and modulates oxidative stress responses. This study aims to investigate the effect and underlying mechanism of TRIM32 silencing on methamphetamine (MA)-induced oxidative stress injuries. PC12 cells were employed as the experimental model, and TRIM32 expression was knocked down using small interfering RNA (siRNA) transfection. The experimental groups were designated as follows: negative control (NC) +NS, NC+MA, siTRIM32+NS, and siTRIM32+MA. Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) assay demonstrated that TRIM32 silencing attenuated MA-induced apoptosis. Colorimetric assay and 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) fluorescent probe detection revealed that TRIM32 silencing reversed MA-induced decreases in superoxide dismutase (SOD) and catalase (CAT) activities (P<0.01), as well as elevations in malondialdehyde (MDA) and reactive oxygen species (ROS) levels (P<0.01). Western blotting analysis indicated that, compared with the NC+MA group, the siTRIM32+MA group exhibited upregulated protein expression of phosphatidylinositol 3-kinase (PI3K) (P<0.05) and phosphorylated protein kinase B (p-AKT) (P<0.01). No significant difference was observed in total nuclear factor erythroid 2-related factor 2 (Nrf2) protein expression. However, the siTRIM32+MA group displayed increased p-Nrf2/Nrf2 ratio (P<0.05) and nuclear Nrf2 (n-Nrf2) protein expression (P<0.01), concomitant with decreased cytoplasmic Nrf2 (c-Nrf2) protein expression (P<0.01) relative to the NC+MA group. Upon simultaneous treatment with the PI3K/AKT inhibitor LY294002, the siTRIM32+MA+LY294002 group showed elevated ROS fluorescence intensity (P<0.05) and MDA levels (P<0.01), reduced SOD expression (P<0.01), unaltered total Nrf2 protein expression, decreased p-Nrf2/Nrf2 ratio and nuclear Nrf2 protein expression (P<0.01), and increased cytoplasmic Nrf2 protein expression (P<0.01) compared with the siTRIM32+MA group. Collectively, these findings demonstrate that TRIM32 silencing exerts neuroprotective effects by suppressing MA-induced oxidative stress injury, potentially through modulation of the PI3K/AKT/Nrf2 signaling pathway.
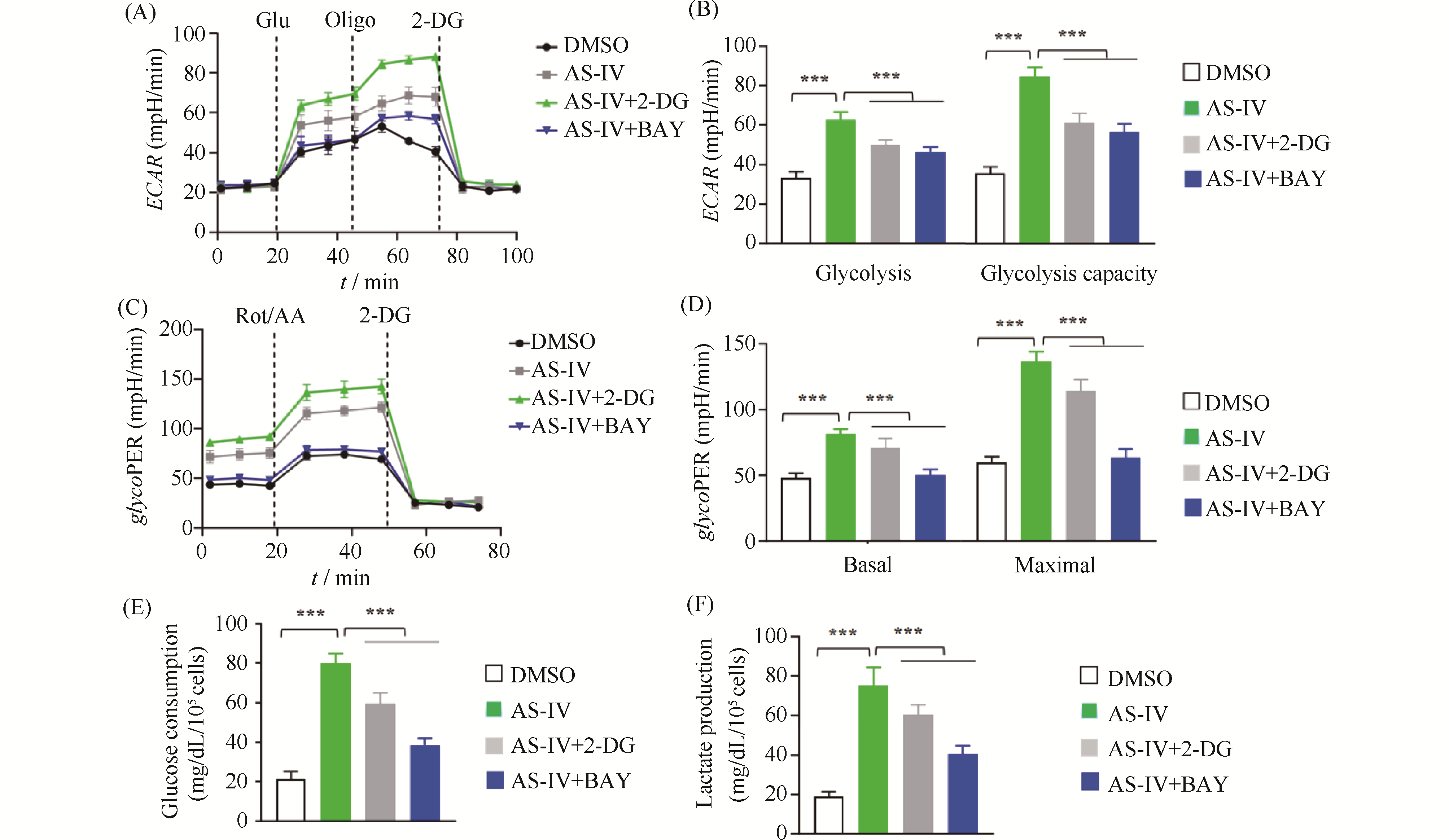
Myocardial ischemia-reperfusion injury is a critical clinical problem in the treatment of cardiovascular diseases, and its pathological process aligns with the traditional Chinese medicine (TCM) pathogenesis of "qi deficiency and blood stasis" as well as "heart vessel obstruction." Modern integrative medicine research suggests that the scientific connotation of "qi" in TCM encompasses oxygen, nutrients, and energy substances such as adenosine triphosphate (ATP). Among these, efficient ATP synthesis via mitochondrial oxidative phosphorylation serves as the primary metabolic basis for a state of abundant "qi," whereas glycolysis acts as a compensatory pathway to maintain basal energy supply under hypoxic conditions. Astragalus membranaceus is a key herb for replenishing qi and activating blood circulation, and Astragaloside IV (AS-IV) is its main active component responsible for cardioprotective effects. However, the regulatory mechanisms by which AS-IV modulates cellular energy metabolism and epigenetic modifications require further elucidation. This study is grounded in the TCM theory of "qi and blood". We established hypoxia/reoxygenation (H/R) injury models using H9c2 rat cardiomyocytes and primary neonatal rat cardiomyocytes. CCK-8 assay, flow cytometry, Seahorse metabolic analysis, Western blotting, and qPCR were employed to investigate the protective effects of AS-IV and its underlying mechanisms. Results showed that pretreatment with 20 μmol/L AS-IV significantly increased cardiomyocyte viability (1.2-fold compared to the H/R model group, P < 0.001) and reduced the apoptosis rate by approximately 47% (P < 0.001). These protective effects were reversed by the glycolysis inhibitor 2-DG or the HIF-1α inhibitor BAY. AS-IV effectively counteracted H/R-induced glycolysis suppression, increasing extracellular acidification rate, glycolytic proton efflux rate, and lactate production (all P < 0.001). Meanwhile, AS-IV treatment not only upregulated HIF-1α expression, but also markedly enhanced its lysine lactylation level (P < 0.001), thereby driving significant upregulation of downstream key glycolytic genes such as SLC2A1, HK2, PDK1, and LDHA at both mRNA and protein levels(mRNA expression level,P < 0.001). These core findings were validated in primary neonatal rat cardiomyocytes. Under hypoxic stress, AS-IV did not restore the high-efficiency pathway of oxidative phosphorylation; instead, it maintained the compensatory energy supply through glycolysis, helping cardiomyocytes survive the energy crisis. This mode of action—"preserving the root of qi"—reflects the TCM therapeutic principle: "Retain a fraction of primordial qi, and then a chance remains for vitality." In conclusion, AS-IV alleviates H/R injuries in cardiomyocytes by promoting HIF-1α lactylation, enhancing its transcriptional activity, and driving glycolytic metabolic reprogramming. This study reveals a novel mechanism of AS-IV's cardioprotective effect from the “metabolic-epigenetic” regulatory axis and provides a modern biological interpretation for the TCM theory of “tonifying Qi and promoting blood circulation.”
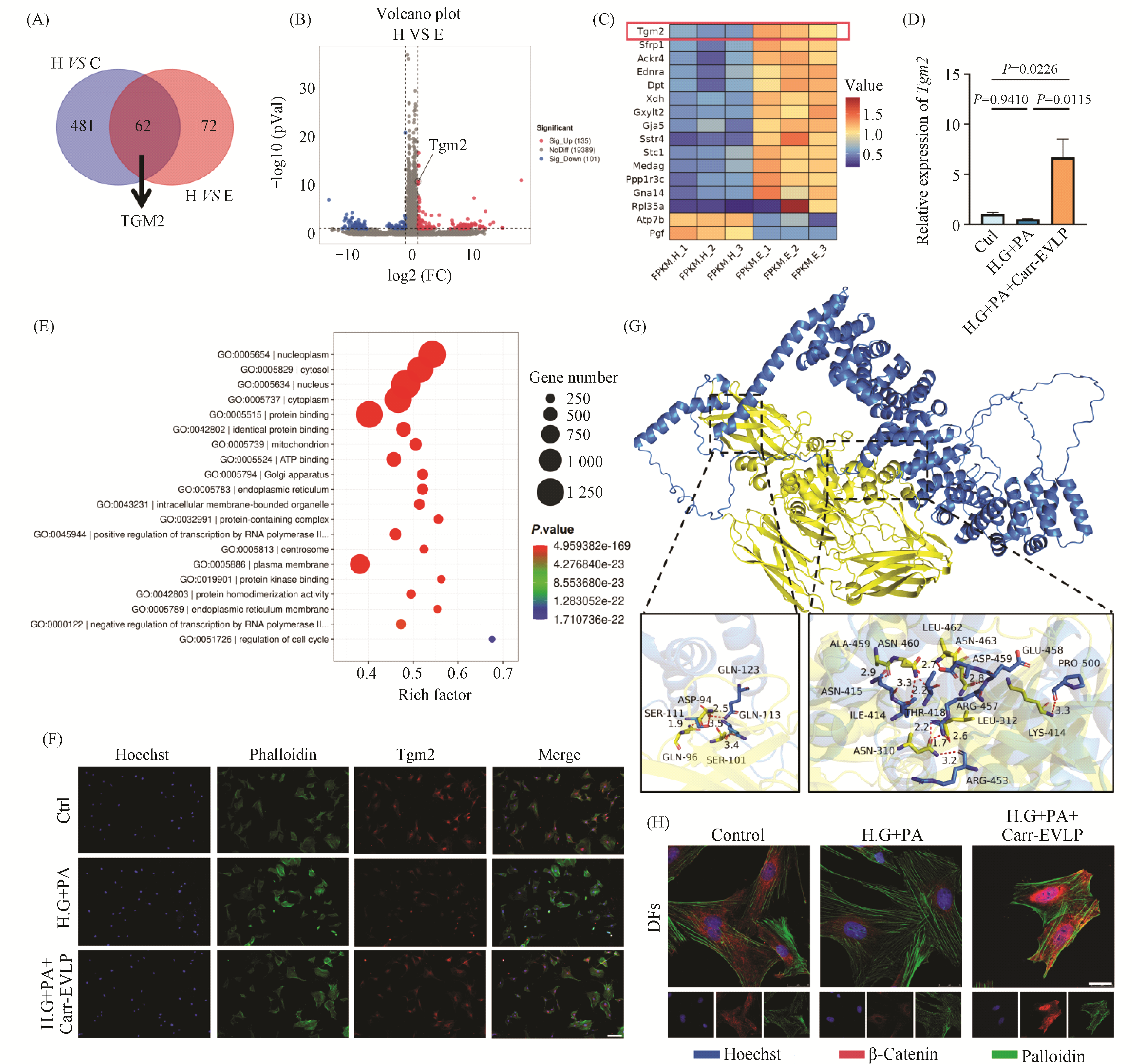
Diabetic wound healing represents a significant clinical challenge, characterized by impaired dermal fibroblast (DFs) function and compromised angiogenesis. Plant-derived extracellular vesicle-like particles (EVLPs), known for their rich bioactive components and good biocompatibility, have garnered increasing attention in regenerative medicine. This study aims to investigate the effects and underlying mechanisms of carrot-derived EVLP (Carr-EVLP) on diabetic wound healing, with a focus on their regulatory roles in DFs and human umbilical vein endothelial cells (HUVECs). The Carr-EVLPs were isolated and characterized by transmission electron microscopy, nanoparticle tracking analysis, and surface marker detection. Under high-glucose and high-lipid conditions, Carr-EVLP treatment significantly reduced the apoptosis rate of DFs and HUVECs (P <0.0001), and enhanced cell proliferation (P <0.0001), migration (P <0.05), and tube formation ability. To further investigate the underlying mechanisms, transcriptomic sequencing was performed and identified transglutaminase 2 (Tgm2) as a potential key mediator. The expression of Tgm2 was subsequently verified by qRT-PCR (P <0.05) and immunofluorescence staining. Additionally, the antimicrobial activity of Carr-EVLP against the common pathogenic bacteria of diabetic foot ulcer (DFU) was evaluated using A600 measurements with a microplate reader, colony plating, and disk diffusion assays. The results suggest that Carr-EVLP may exert its effects by influencing Tgm2 expression and engaging the β-catenin signaling pathway, thereby synergistically enhancing the functionality of DFs and HUVECs, while also exhibiting antimicrobial properties. These findings provide a novel strategy and theoretical foundation for the treatment of diabetic wounds.
In response to the national strategic call for the development of the "New Agricultural Sciences Initiative" and to address the prevalent issue of disconnection between professional knowledge education and ideological and political education in basic theoretical courses such as "Molecular Biology", this study designed and implemented a new Artificial Intelligence (AI)-empowered teaching model for integrating ideological and political education into the "Molecular Biology" course, targeting undergraduate students majoring in Seed Science and Engineering and Plant Science and Technology at Gansu Agricultural University. This model leverages AI to deeply mine and systematically construct a case library of ideological and political materials that is organically integrated with the knowledge points of molecular biology. Through a four-stage, closed-loop teaching process—"AI-driven inquiry before class, human-computer collaborative exploration during class, AI-assisted consolidation after class, and data-driven reflection throughout"—the model integrates value guidance into the entire process of knowledge transmission. Empirical data demonstrates that this model significantly enhanced comprehensive teaching effectiveness: in terms of academic performance, the excellence rate (>90 points) in the final exam for the experimental group was 2.6 times that of the control group, while the rates of low scores (60-69) and failures were both halved. Moreover, in the "Simulated Academic Presentation" segment assessing higher-order thinking skills, the experimental group significantly outperformed the control group (p<0.05). In terms of learning experience, 94.85% of the experimental group students found the integration of ideological and political cases natural and inspiring, reporting a significantly enhanced professional identity in serving the agricultural sector. The findings indicate that the AI-empowered teaching model can effectively solve the challenge of integrating ideological and political education into theoretical courses, aligning knowledge transmission with value shaping. It provides a replicable and scalable practical solution for cultivating innovative talents in the new era who possess solid professional skills, a deep sense of patriotism and national pride, and a strong understanding of and affection for agriculture.
Monthly journal, established in 1985 Sponsored by:
Chinese Society of Biochemistry and Molecular Biology
Peking University Undertaken by:
Peking University Health Science Center Edited by:
Editorial Office of Chinese Journal of Biochemistry and Molecular Biology Editor-in-Chief:
CHANG Zheng-Yi
ISSN 2097-4329 (Online)
ISSN 1007-7626 (Print)
CN 11-3870/Q