ZHAO Lan , ZHANG Meng , WU Ling , YAN Guang-Dong , YU Meng-Ying , ZHANG Shao-Heng
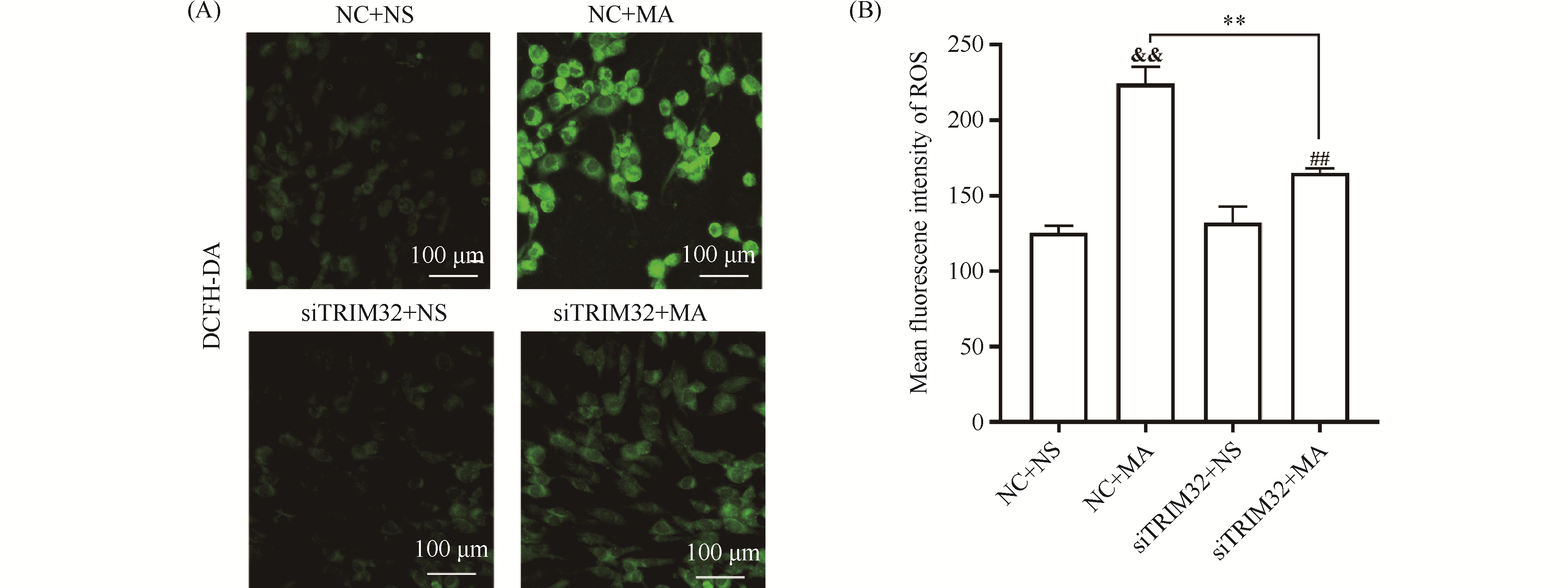
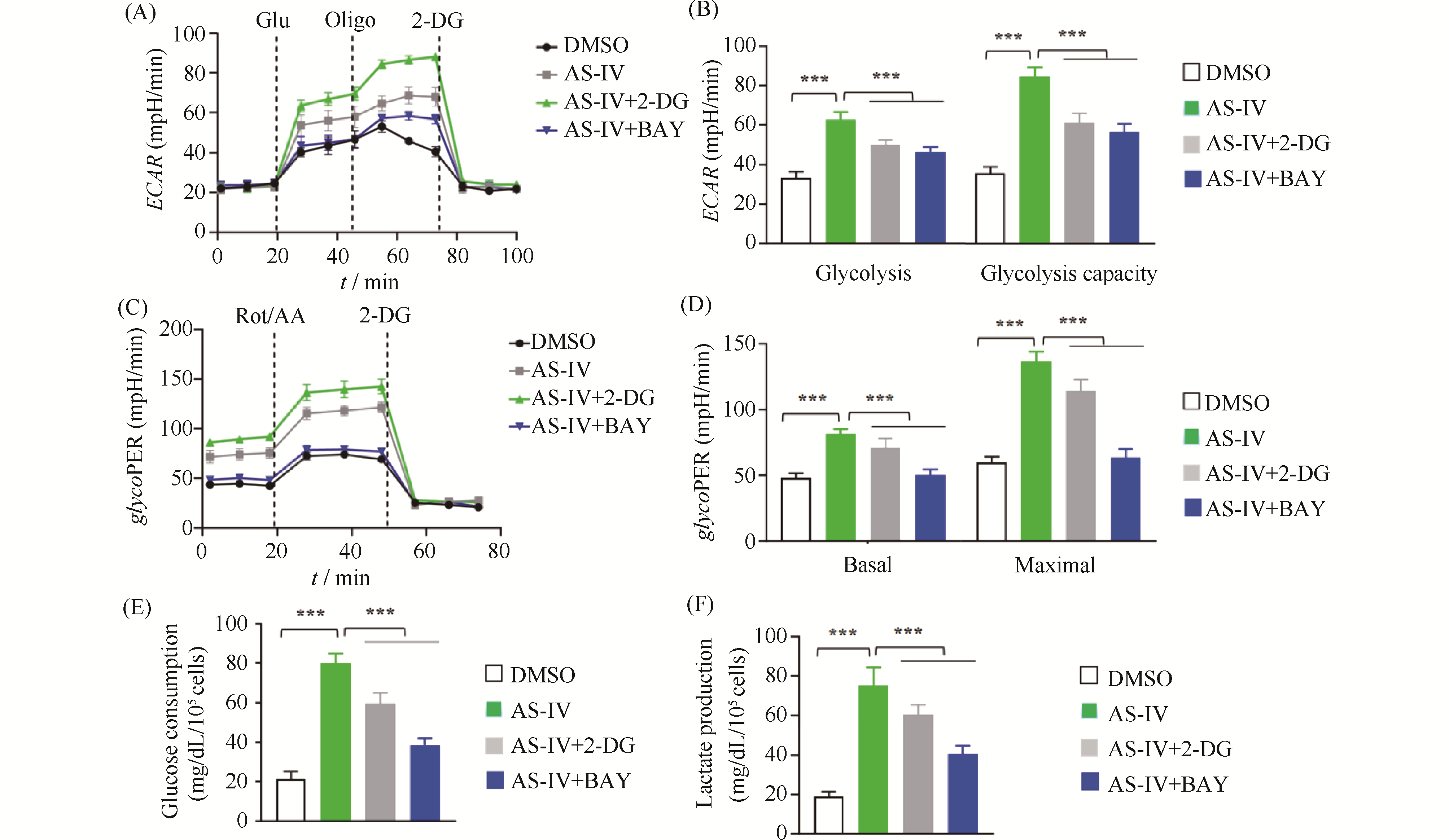
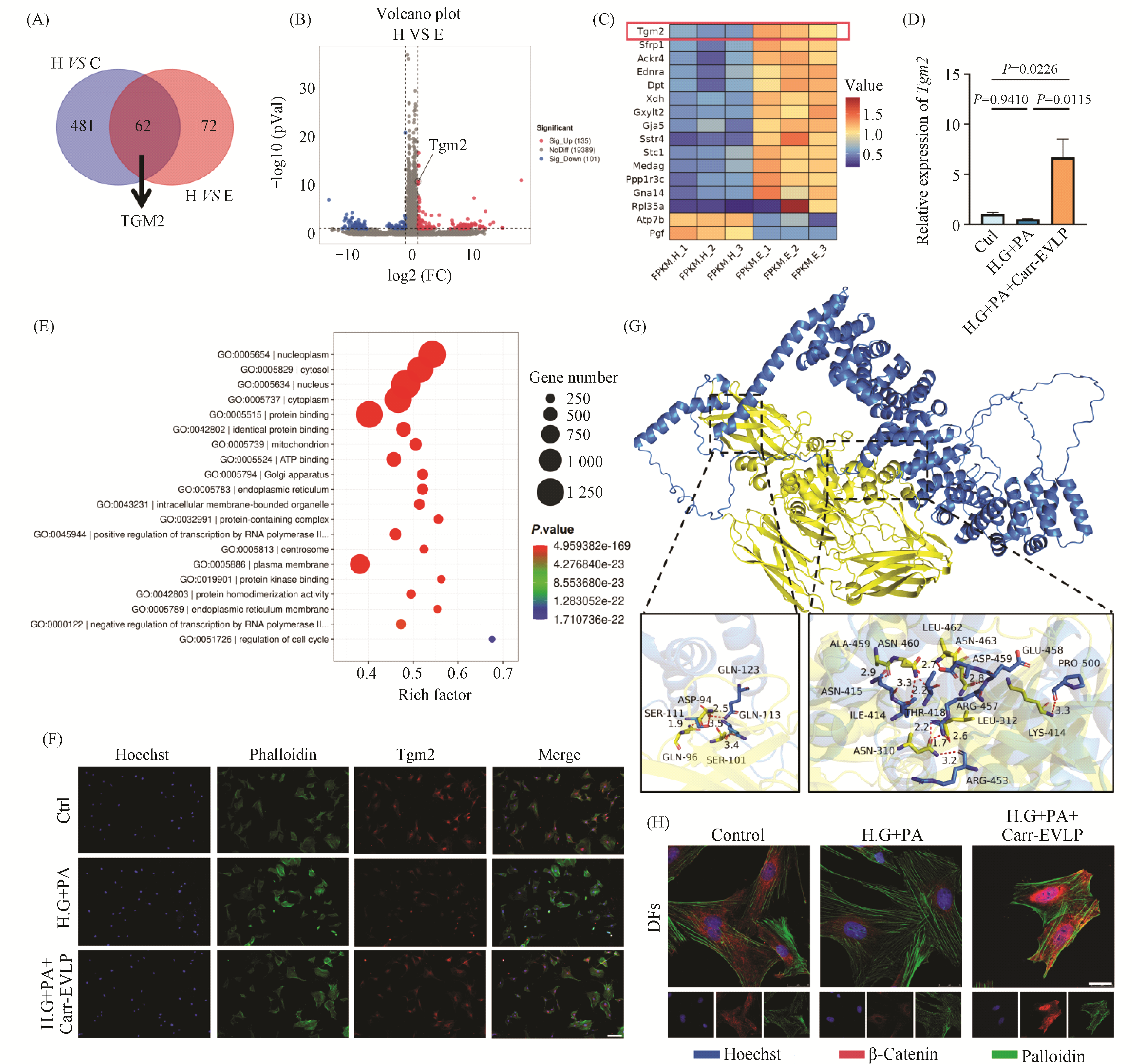
Myocardial ischemia-reperfusion injury is a critical clinical problem in the treatment of cardiovascular diseases, and its pathological process aligns with the traditional Chinese medicine (TCM) pathogenesis of "qi deficiency and blood stasis" as well as "heart vessel obstruction." Modern integrative medicine research suggests that the scientific connotation of "qi" in TCM encompasses oxygen, nutrients, and energy substances such as adenosine triphosphate (ATP). Among these, efficient ATP synthesis via mitochondrial oxidative phosphorylation serves as the primary metabolic basis for a state of abundant "qi," whereas glycolysis acts as a compensatory pathway to maintain basal energy supply under hypoxic conditions. Astragalus membranaceus is a key herb for replenishing qi and activating blood circulation, and Astragaloside IV (AS-IV) is its main active component responsible for cardioprotective effects. However, the regulatory mechanisms by which AS-IV modulates cellular energy metabolism and epigenetic modifications require further elucidation. This study is grounded in the TCM theory of "qi and blood". We established hypoxia/reoxygenation (H/R) injury models using H9c2 rat cardiomyocytes and primary neonatal rat cardiomyocytes. CCK-8 assay, flow cytometry, Seahorse metabolic analysis, Western blotting, and qPCR were employed to investigate the protective effects of AS-IV and its underlying mechanisms. Results showed that pretreatment with 20 μmol/L AS-IV significantly increased cardiomyocyte viability (1.2-fold compared to the H/R model group, P < 0.001) and reduced the apoptosis rate by approximately 47% (P < 0.001). These protective effects were reversed by the glycolysis inhibitor 2-DG or the HIF-1α inhibitor BAY. AS-IV effectively counteracted H/R-induced glycolysis suppression, increasing extracellular acidification rate, glycolytic proton efflux rate, and lactate production (all P < 0.001). Meanwhile, AS-IV treatment not only upregulated HIF-1α expression, but also markedly enhanced its lysine lactylation level (P < 0.001), thereby driving significant upregulation of downstream key glycolytic genes such as SLC2A1, HK2, PDK1, and LDHA at both mRNA and protein levels(mRNA expression level,P < 0.001). These core findings were validated in primary neonatal rat cardiomyocytes. Under hypoxic stress, AS-IV did not restore the high-efficiency pathway of oxidative phosphorylation; instead, it maintained the compensatory energy supply through glycolysis, helping cardiomyocytes survive the energy crisis. This mode of action—"preserving the root of qi"—reflects the TCM therapeutic principle: "Retain a fraction of primordial qi, and then a chance remains for vitality." In conclusion, AS-IV alleviates H/R injuries in cardiomyocytes by promoting HIF-1α lactylation, enhancing its transcriptional activity, and driving glycolytic metabolic reprogramming. This study reveals a novel mechanism of AS-IV's cardioprotective effect from the “metabolic-epigenetic” regulatory axis and provides a modern biological interpretation for the TCM theory of “tonifying Qi and promoting blood circulation.”


{kind=link}